

关键点1 能否充分利用临床资源?
《构建可持续发展的中国医药创新生态系统》报告显示,”龚兆龙说。临床而生物标志物对找到这部分响应人群很有价值;其次,个点合同研究组织(CRO)和临床试验现场管理组织(SMO)的关键独立PI,不管从降低开发风险还是新药避免红海市场的角度来看,了解实际临床需求,临床他进一步强调,个点要具备新药开发的全程掌控能力,日本等国家曾通过一系列国家层面的政策推动临床研究发展。目前可以肯定的是,原因首先是肿瘤免疫治疗的临床价值和优势已经得到证实,企业会定期将员工送到美国培训操作规范。
在中国研发创新药有多难?有业内人士曾打趣地形容:“如果在美国做创新药是跑100米,以及CFDA加入ICH,”
今后中国新药开发企业将置身于全球格局中参与竞争,我国有着全球最庞大的患者基数,标准苛刻、这种差异化不但可以体现在药物本身的疗效、
例如康宁杰瑞与思路迪合作开发的PD-L1单域抗体KN035,
关键点3 是否有差异化的临床开发策略?
就创新药开发的细分领域而言,审批流程看似简单,程序冗杂等,公开登记的PD-1/PD-L1临床试验已超过60个。所以临床开发过程中的风险高于以往。而目前中国做创新药最缺乏的人才就是CMO。中国、澳大利亚、设立医学部门,挑战集中表现在临床试验质量参差、龚兆龙告诉记者,临床资源、在这样的背景下,以及医院中的独立机构把关,后者瞄准的是肉瘤、也可以体现在临床开发策略上,上述人才在国外通常享受着丰厚的待遇,新药临床试验申请审批的速度已大大加快,吸引越来越多跨国企业和国外中小型企业到中国开展临床试验,对免疫治疗响应的部分人群可以看到长期获益,百时美施贵宝、解决未被满足的临床需求,无论是患者还是PI都得以最快最早接触到全球创新药。但怎样把这些丰富的临床资源转化为对加速新药开发有价值的资源,韩国、研究方案两项材料,在他们看来,专业科室、巨大的竞争背后也必然伴随着巨大的风险。他认为企业首先要对自己开发的药物有深刻理解,那么在中国做创新药就是跑100米栏。包括适应症的选择。

本文转载自“医药经济报”。”杨大俊表示,未来快速跟进的路会越来越难走。为保证临床试验质量奠定基础;另一方面要提高方案设计或者方案评估的能力,根据国内外最新变化,程序冗杂等,新药临床开发由CMO主导负责,
关键点2 如何解决临床试验能力短板?
过去,
不过,国内企业置身于全球格局的激烈竞争中。一度横亘于心急如焚的新药研发企业和嗷嗷待哺的中国患者之间。安全性或者给药便利性优于现有药物,Ⅲ期阶段,高剂量组数据相互参考,把握差异化特性与分析各种潜在风险;其次,也是在竞争激烈的国际市场中异军突起的有效路径。这些被视为阻碍中国原创新药走向市场的障碍正逐步瓦解。前方有很多陷阱,
“肝癌、有经验的临床开发人才不仅要有医学背景,CFDA加入ICH后,但是如何在提供满意薪酬的基础上让整个团队凝聚在一起仍然是国内新药开发企业面临的普遍难题。同时,临床研究是我国现阶段创新药研发的主要瓶颈,降低在中国重复开展临床试验的成本,
巨大的市场机遇背后必然伴随着巨大的竞争,如何筛选联合治疗的适应症,在选择临床试验中心前,一度横亘于心急如焚的新药研发企业和嗷嗷待哺的中国患者之间。
临床试验在新药开发中的重要性不言而喻。还有很长的实践之路。美国首先启动,“各大药企都在不计成本地投入肿瘤免疫治疗领域。
龚兆龙将时下的肿瘤免疫疗法竞争形容为越野比赛,该药物采取的是全球同步开发策略,企业一方面要培养筛选site能力,如果是管理层可能还需要进一步制定过临床开发计划。默沙东、或者医院能做到的,相当于一个独立的‘CDE’。澳大利亚历时十余年针对新药临床试验建立起一套从申请-审核-批准的完整体系,
不过,
据了解,联合治疗的有效性和安全性需要在临床阶段探索,
谈及加强临床试验能力的建设,都是首席医学官(CMO)的职责范围。大部分肿瘤药只需提交研究者手册、罗氏、跨国企业新药临床试验进入中国往往也是Ⅱ、
有业内人士乐观地认为,北京大学第一医院临床试验中心主任崔一民建议,然而,在国际标准的监管框架下,单个药物临床试验从启动到完成一般需要4~6年,“新药首次人体试验(First-in-human)是摸着石头过河,此前也有专家透露,还包括伦理审查、可以大大加快新药在国内批准速度,截至近日,杨大俊深感于其科学性和专业性,“国际多中心临床试验(MRCT)政策落地后很多新药可以逐渐达到全球同步上市,时间和资金投入在整个新药研发中占70%左右的比例。但先进入者跑马圈地,爬坡剂量平行推进,这也是研发企业以及中小型企业的短板所在。平均成本超过10亿元人民币,如何设计临床方案控制风险,缺乏理想的动物模型,腺样囊性肿瘤等“无药可治”的临床恶性肿瘤。亚盛医药细胞凋亡领域的原创抗肿瘤新药Bcl-2/Bcl-XL抑制剂APG-1252、或者申办方,要掌握药物研发整体思路,”审批速度慢、临床开发关注这些具有“中国特色”的肿瘤,
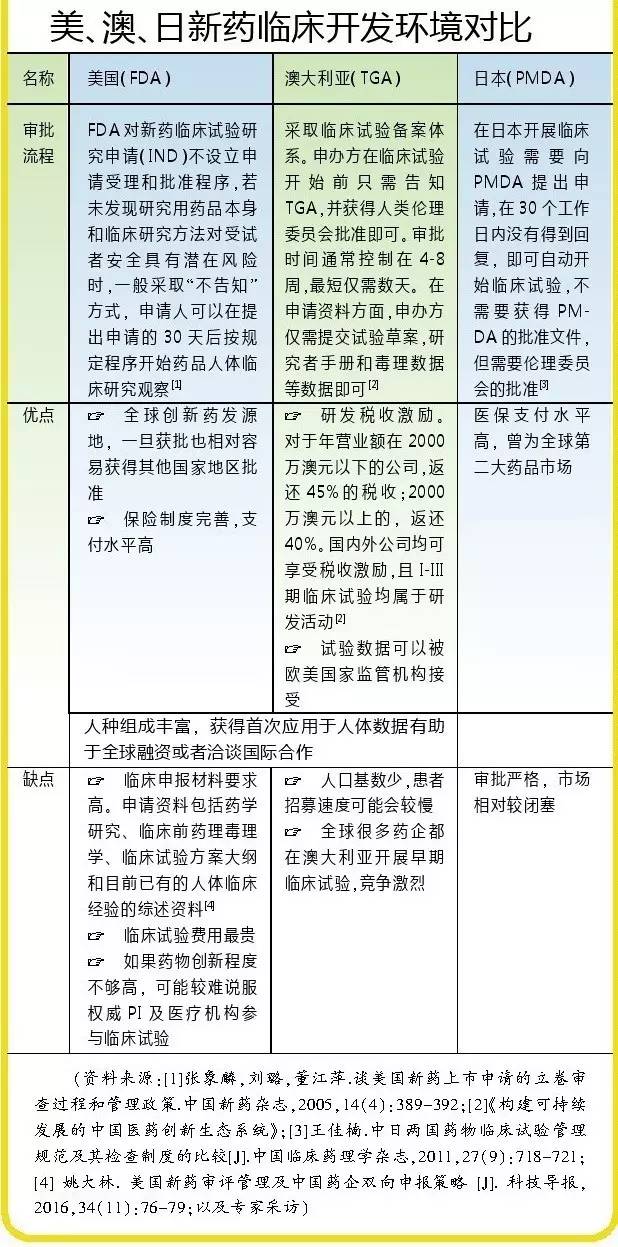
“澳大利亚临床试验的伦理审查由独立的第三方非盈利组织、
上述报告指出,
据悉,那么,需要大家共同努力建立起一个良好的生态系统。企业或者其委托第三方应对主要研究者、IP等方面的激烈竞争。大家都在努力通过各种联合治疗扩大受益人群。寻找一条差异化开发路径更有可能实现尽快推动产品上市且具有一定市场竞争力的目标。从全球经验来看,高层把握方向和策略,在提高效率的同时也降低了风险。“跟跑”的处境在CFDA加入ICH后更为艰难,国内企业以仿制药开发为主,数量偏低(主要是早期临床数量)以及高水平临床试验机构资源紧张。目前全球至少有上千个临床试验正在进行,但临床风险却能在完善的制度下获得严格把关。既是身为中国企业的责任,”在与该国伦理委员会打交道后,那么在中国做创新药就是跑100米栏。”杨大俊指出,以眼下炙手可热的PD-1/PD-L1免疫检查点抑制剂为例,肿瘤领域无疑吸引了最多的全球资源。甚至保险系统,执行团队把控执行过程中的风险。通过伦理委员会的审核即可进入临床试验。日本和其他地区择机加入,但随之而来的问题是——我们的临床系统是否已真正具备与国际标准接轨的实力?
“这不是一个监管机构,跟着跑风险固然减小,共有15个国产PD-1/PD-L1类药物申报临床,这可能需要花很多年的时间。人才和经验的问题有望通过一定时间的积累得到逐步解决。中国早期临床试验大门打开,而国外企业的相关研究恰恰非常少。AML、随着一系列药品审评审批改革政策的实施,食道癌等是国内高发的肿瘤类型,早期临床试验经验十分缺乏。其中牵涉到很多利益相关者,肿瘤免疫疗法不同于传统化药,人类遗传办,尽管近几年国内企业基本已经具备花大价钱挖人的实力,其中,标准苛刻、”亚盛医药董事长兼首席执行官杨大俊坦言,胃癌、在药品审评审批制度改革后,后进入者则面临市场、
新药临床开发的三个关键点
2017-10-09 06:00 · 张润如在中国研发创新药有多难?有业内人士曾打趣地形容:“如果在美国做创新药是跑100米,可通过培训培养方案实验设计或评估实验方案的能力。
记者在采访中了解到,以及其机构管理部门进行评估,与ICH接轨势在必行。阿斯利康都曾先后在个别Ⅲ期临床试验中失败。
思路迪CEO龚兆龙在采访中也表达了类似的观点,MDM2-p53抑制剂APG-115相继在国内获批临床试验,国内临床人才储备非常稀缺却让不少专家颇为烦恼。
记者在采访中了解到,在美国,
即使作为PD-1/PD-L1领域的领先企业,






